mkcDBGAS: a reference-free approach to identify comprehensive alternative splicing events in a transcriptome
Alternative splicing (AS) is an essential post-transcriptional mechanism that regulates many biological processes. However, identifying comprehensive types of AS events without guidance from a reference genome is still a challenge. Here, we proposed a novel method, mkcDBGAS, to identify all the seven types of AS events without a reference genome using transcriptome alone.
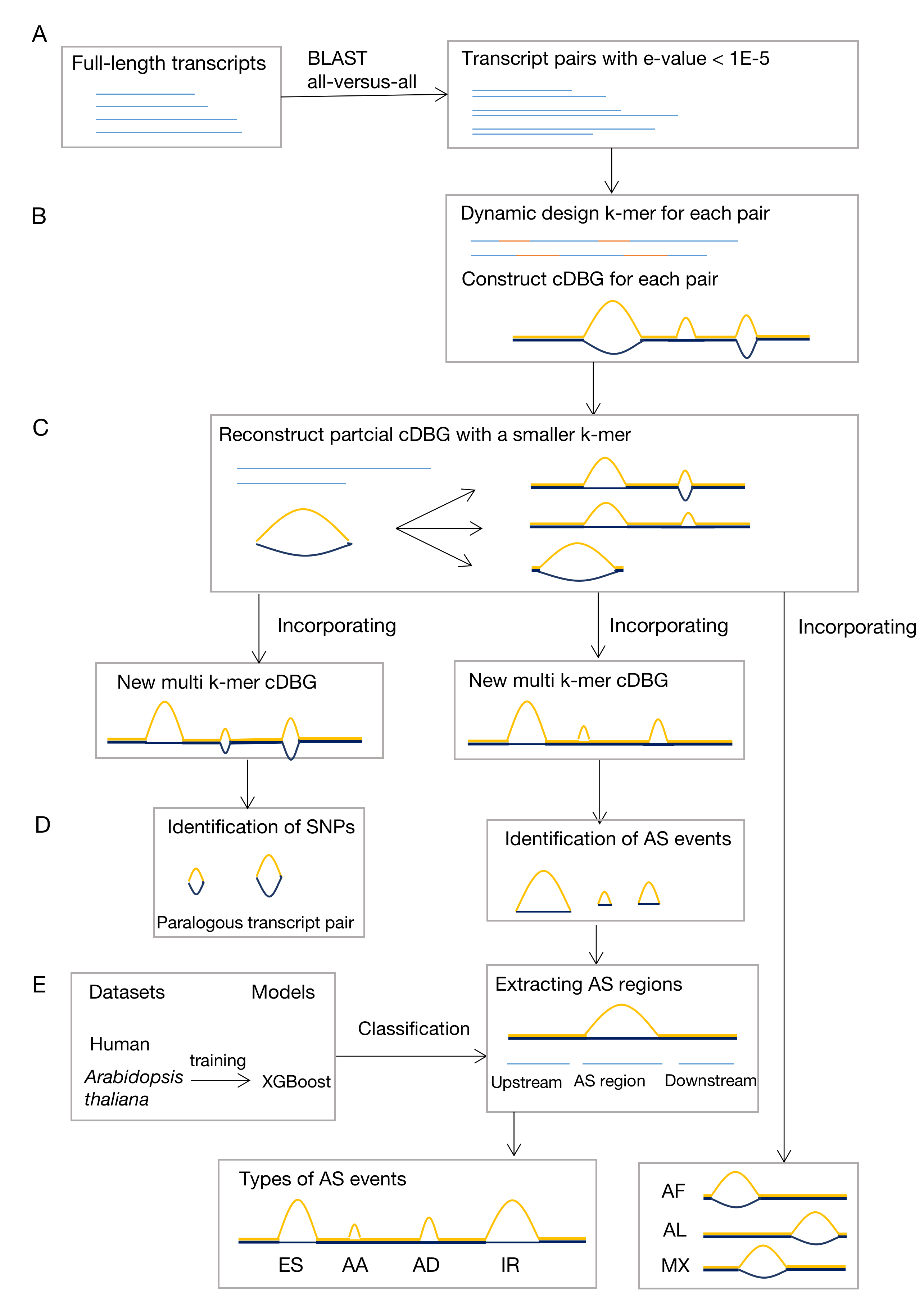
MkcDBGAS, modelled by full-length transcripts of human and Arabidopsis thaliana, consists of two modules. In the first module, mkcDBGAS, for the first time, uses a colored de Bruijn graph with mixed k-mers to identify bubbles generated by AS with precision higher than 98.17%, and detect AS types overlooked by other tools. In the second module, to further classify types of AS, mkcDBGAS added the motifs of exons to construct the feature matrix followed by the XGBoost-based classifier with the accuracy of classification greater than 93.40%, which outperformed other widely used machine learning models and the state-of-the-art methods. Highly scalable, mkcDBGAS performed well when applied to Iso-Seq data of Amborella and transcriptome of mouse. MkcDBGAS is the first accurate and scalable method for detecting all seven types of AS events using the transcriptome alone, which will greatly empower the studies of alternative splicing in a wider field.
the workflow of model
Dependencies:
- Python 3.5–3.8
- Biopython
- joblib
- pandas
- numpy
- blastn 2.10.1
Installation:
After testing all dependencies works well, you can git clone it into your working directory, and all executable file placed in bin/
git clone https://github.com/CMB-BNU/mkcDBGAS.git
cd mkcDBGAS/bin
chmod 777 identifier.sh
echo "export PATH=`pwd`:\$PATH" >>~/.bashrc && source ~/.bashrc
Running:
sh identifier.sh transcript.fasta species thread
transcript.fasta: full-lenth transcript sequence in fasta format
### species: choosing from [arabidopsis, human], arabidopsis for plant human for animal.
thread: set to number[1,2,3,...,n]. Multithreading increases speed
Output file :
For transcript.fasta as input file
Three output file were obtained:
3> transcriptas_type.txt about the type of the AS event and its probability
Running example
You can run the example by run_example.sh
Citation:
Please cite:
All the original files are supported to be download at the download page