DeepASmRNA: attention-based convolutional neural network method with scalability and interpretability for predicting alternative splicing events from transcript sequences without a reference genome
The sharp increase in the number of sequenced transcriptomes without reference genomes has empowered the investigation of AS events. We proposed an attention-based CNN model, DeepASmRNA, an accurate, scalable and biologically interpretable tool for predicting AS events using a transcriptome without a reference genome, which is a step towards investigating AS genome-wide in species without a reference genome. To our knowledge, DeepASmRNA is the first only dependence of primary sequences of mRNA transcripts for predicting AS at the genome-wide level. DeepASmRNA will greatly expand the studies of alternative splicing in species without a reference genome.
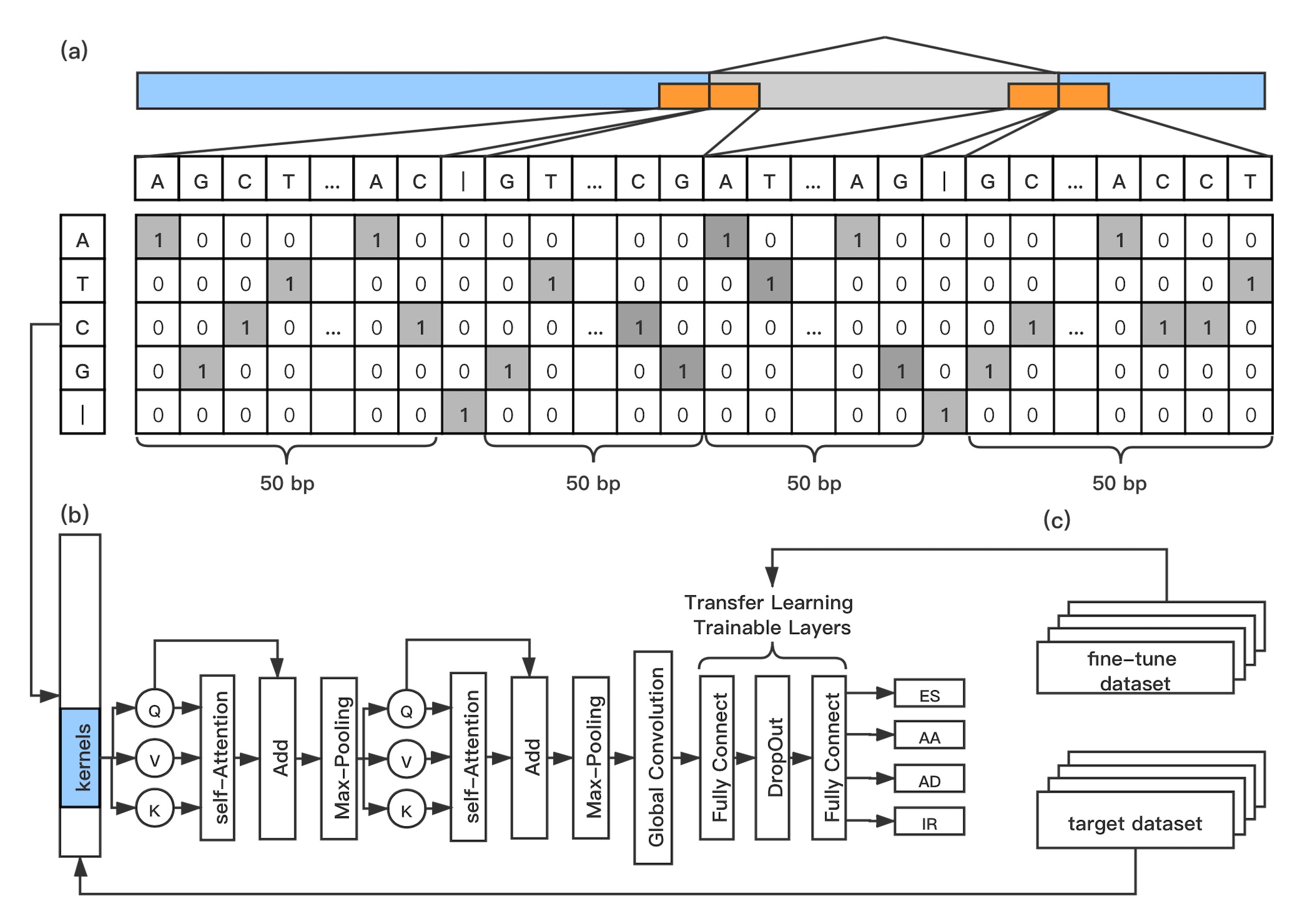
Our method, DeepASmRNA, is composed of two parts. For the first part, we use all-versus-all BLASTN to identify alternatively spliced transcripts. For the second part, we use an attention-based convolution neural network (CNN) approach to classify 4 basic types of AS: exon skipping (ES), alternative acceptor site (AA), alternative donor site (AD) and intron retention (IR). Our model takes the primary sequence of transcripts without a reference genome as input and outputs the probability of ES, AA, AD and IR for each transcript pair.
the workflow of model
Dependencies:
- Python 3.5–3.8
- TensorFlow >2.1
- blastn 2.10.1
We strongly recommend using Anaconda to install all dependencies. You can simply install the dependencies by running following commands.
conda create -n DeepASmRNA python=3.7
conda activate DeepASmRNA
conda install tensorflow=2.1
conda install biopython
Installation:
After testing all dependencies works well, you can git clone it into your working directory, and all executable file placed in bin/
git clone https://github.com/CMB-BNU/DeepASmRNA.git
cd DeepASmRNA/bin
chmod 777 identifier.sh
echo "export PATH=`pwd`:\$PATH" >>~/.bashrc && source ~/.bashrc
Running:
first way: for overall workflow
conda create -n DeepASmRNA python=3.7
conda activate DeepASmRNA
sh identifier.sh transcript.fasta species ### species, choosing from [ara, human], ara for plant human for animal
second way: step by step
1). predict AS event
For predict AS event:
makeblastdb -in transcript.fasta -dbtype nucl ### make blast database
blastn -query transcript.fasta -db transcript.fasta -strand plus -evalue 1E-10 -outfmt 5 -ungapped -num_threads 20 -out transcript.xml ### sequence alignment using blastn
python3 predictAS.py transcript.xml transcriptas.txt >transcript.seq ### predict AS transcript pair
2). classify AS event
You can run AS classification model by
python classifyAS.py transcript.seq \ ### input file name
-m human \ ### optional, model for species, choosing from [ara, human, rice, fine_tune], default = human
-o transcriptas_type.txt ### optional, output file name
Also you can use a very small dataset to enhance the performance of model, for example:
python classfyAS.py PATH/TO/input_file \ ### input file name
-m human \ ### optional, model for species, choosing from [ara, human, rice, fine_tune], default = human
-o PATH/TO/input_file \ ### optional, output file name
-ft PATH/TO/fine_tune_input_file ### optional, fine tune input file
Different from 'input' option, the 1st column of fine tune file should be the label of the sequence, choosing from (A3, A5, ES, IR).
If you use -ft option to fine tune the model, the meaning of -m is the basic model you choose to be trained, the original model will not be changed after each time of fine tune learning.
Output file :
For transcript.fasta as input file
Three output file were obtained:
1> transcriptas.txt about the AS events information which including position, identity, coverage, length
2> transcriptas.seq about the sequence of AS events which including the AS region and upstream 50 bp and downstream 50 bp
3> transcriptas_type.txt about the type of the AS event and its probability
Running example
You can run the example by run_example.sh
Citation:
Please cite:
All the original files are supported to be download at the download page