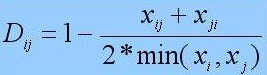 , i, j = 1, 2, ..., N
, i, j = 1, 2, ..., N
BPhyOG is an interactive web server for the reconstruction of whole-genome bacterial phylogenies based on overlapping genes, and it also allows users to browse overlapping genes to acquire more information in the subtree-specific genomes or in the whole set of genomes. Fukuda et al. and Johnson et al. also showed that the evolution of overlapping gene structures may be related to the evolutionary time scale [1-3]. Therefore, assuming a universal rate of formation and degradation of overlapping genes across species, we can determine the evolutionary distance between two bacterial genomes on the basis of the number of their shared overlapping gene pairs [4]. It is a useful tool for analyzing the tree of life and overlapping genes from a genomic standpoint.
Features of overlapping genes
Overlapping genes (OGs) are defined as pairs of adjacent genes of which the coding sequences overlap partly or entirely.
ORF p (q): the individual overlapping gene p (q) in an OG pair; all the features of the individual gene are extracted from the GenBank file from NCBI.
Overlap length: the length of the overlap segment in an OG pair.
Overlap direction: the relative orientation of two individual overlapping genes:ĄŪconvergentĄŊ (-><-), ĄŪunidirectionalĄŊ (->->), and ĄŪdivergentĄŊ (<-->).
Overlap phase type: the corresponding codon positions of the overlap segment takes in two individual overlapping genes, denoted such asĄŪ<2:3, 1:2>ĄŊ.
Orthologous overlapping gene pairs:
Orthologous OG pairs from two different genomes (genome i and genome j) are defined as pairs of genes that overlap in genome i and that have orthologous counterparts that overlap in genome j [4].
The possible orthologous genes among 177 genomes were examined using NCBI BLAST version 2.2.6 [Apr-09-2003, for Linux IA-64 systems] [5] through searching bidirectional best hits and applying thresholds of e-value <10ĻC4 and identity >40%.
Reconstruction of phylogenies
BPhyOG allows users to infer phylogenetic relationships for a set of genomes of interest on the basis of the number of orthologous OG pairs and the inferred tree will be directly visualized online.
Distance measure:
For the phylogenies based on overlapping genes, similarity between two genomes is defined as the ratio of the number of shared orthologs and a normalization value that reflects varying the number of overlapping genes in different genomes. The evolutionary distance between two genomes is defined as:
, i, j = 1, 2, ..., N
where xi is the number of OG pairs in genome i, N the number of selected species and xij is the number of OG pairs in genome i that have their respective orthologs in genome j.
Clustering algorithm:
1. Neighbor-joining [6] is a general clustering algorithm.
2. Unweighted PairĻCGroup Method using Arithmetic averages (UPGMA) [7] is more suitable for inferring phylogenies when the rate of evolution of the phylogenetic marker is relatively constant [8].
Genome features
Tid: taxonomy id for the species defined in NCBI Taxonomy database.
Genome sequence size: the total number of nucleotides in whole genome.
Coding sequence size: the total number of nucleotides of annotated coding genes in whole genome.
No. of ORFs: the number of open reading frames (ORFs) annotated as protein coding genes in whole genome.
No. of overlapping pairs: the number of overlapping gene pairs in whole genome.
How to get started with BPhyOG
Phylogenetic inference
Essential inputs of BPhyOG are (1) the desired reconstruction method (UPGMA or NJ), and (2) a set of species to be included in the phylogeny. If ĄŪphylogeny inference' is chosen, the user can s select a set of species of interest (by checking the boxes) on a new page.
By default, the output format is a JPEG-image of a graphical unrooted tree, with the option to download the tree in Newick format.
OG pairs browse
Users can browse all OG pairs in a genome or a particular OG pair of interest, in two ways: through hyperlinks or by manual searching.
References
1. Johnson ZI, Chisholm SW: Properties of overlapping genes are conserved across microbial genomes. Genome Res 2004, 14(11):2268-2272.
2. Fukuda Y, Nakayama Y, Tomita M: On dynamics of overlapping genes in bacterial genomes. Gene 2003, 323:181-187.
3. Fukuda Y, Washio T, Tomita M: Comparative study of overlapping genes in the genomes of Mycoplasma genitalium and Mycoplasma pneumoniae. Nucleic Acids Res 1999, 27(8):1847-1853.
4. Luo Y, Fu C, Zhang DY, Lin K: Overlapping genes as rare genomic markers: the phylogeny of gamma-Proteobacteria as a case study. Trends Genet 2006, 22(11):593-596.
5. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25(17):3389-3402.
6. Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987, 4(4):406-425.
7. Sokal R, and P. Sneath.: Numerical Taxonomy. San Francisco.: Freeman Press; 1973.
8. Nei M, Kumar S: Molecular Evolution and Phylogenetics: Oxford University Press; 2000.